Пиридин
Энциклопедия Брокгауза Ф.А. и Ефрона И.А. (1890 - 1916гг.) Статьи для написания рефератов, курсовых работ, научные статьи, биографии (118447 статей и 6000 рисунков).
|
|
| А | Б | В | Г | Д | Е | Ё | Ж | З | И | Й | К | Л | М | Н | О | П | Р | С | Т | У | Ф | Х | Ц | Ч | Ш | Щ | Ы | Э | Ю | Я | A | B | C | D | E | F | G | H | I | J | K | L | M | N | O | P | Q | R | S | T | U | V | W | Z |
| П | ПЕ | ПА | ПЕ | ПЁ | ПЖ | ПИ | ПЛ | ПН | ПО | ПП | ПР | ПС | ПТ | ПУ | ПФ | ПХ | ПЧ | ПШ | ПЫ | ПЬ | ПЭ | ПЮ | ПЯ |
| ПИА |
| ПИБ |
| ПИВ |
| ПИГ |
| ПИД |
| ПИЕ |
| ПИЖ |
| ПИЗ |
| ПИИ |
| ПИЙ |
| ПИК |
| ПИЛ |
| ПИМ |
| ПИН |
| ПИО |
| ПИП |
| ПИР |
| ПИС |
| ПИТ |
| ПИУ |
| ПИФ |
| ПИХ |
| ПИЦ |
| ПИЧ |
| ПИШ |
| ПИЩ |
| ПИЭ |
| ПИЯ |
Пиридин С 5 Н 5 N. — Это вещество вместе с целым рядом аналогичных ему соединений, вообще называемых пиридиновыми основаниями, было открыто в 1846 г. Андерсоном при исследовании костяного масла, получающегося сухой перегонкой необезжиренных костей. С момента своего открытия Пиридин весьма заинтересовал химиков своими свойствами, во многом напоминающими свойства бензола, и тем, что начинал собой целый ряд гомологов. В 1869 г. Кернер в частном письме к Каниццаро высказал мысль, что Пиридин может быть рассматриваем, как бензол, в котором одна группа СН замещена азотом, т. е. что Пиридин имеет строение: 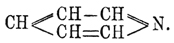 . По мнению Кернера, подобная формула не только объясняет синтезы Пиридин; но, главным образом, указывает, почему простейший член ряда пиридиновых оснований имеет пять атомов углерода. Через год Дьюар (Dewar), независимо от Кернера, пришел к той же формуле, которая затем нашла себе подтверждение и в позднейших работах друг. химиков. В этом отношении особенно интересен синтез Ладенбурга, который из пентаметилендиамина получил пиперидин (см. ниже), легко окисляющийся в Пиридин:
. По мнению Кернера, подобная формула не только объясняет синтезы Пиридин; но, главным образом, указывает, почему простейший член ряда пиридиновых оснований имеет пять атомов углерода. Через год Дьюар (Dewar), независимо от Кернера, пришел к той же формуле, которая затем нашла себе подтверждение и в позднейших работах друг. химиков. В этом отношении особенно интересен синтез Ладенбурга, который из пентаметилендиамина получил пиперидин (см. ниже), легко окисляющийся в Пиридин:
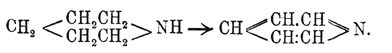
Этот синтез с несомненностью установил замкнутое строение Пиридин, что же касается расположения двойных связей, то вопрос этот оставался открытым, да и до сих пор не решен вполне удовлетворительно. Здесь мы, быть может, встречаемся с тем же фактом, который наблюдается для бензола, ацетоуксусного эфира, нитрозофенолов и многих других соединений, именно: по-видимому, взаимное положение двойных связей Пиридин не постоянно и изменяется в различных его производных. Есть много данных за формулу Кернера и Дьюара; однако в 1883 г. Либен и Гайтингер, с одной стороны, и Ридель, с другой, пришли для Пиридин к формуле 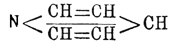 . Несколько ранее (1881 г.) Чамичан (Ciamician) и Деннштедт дали Пиридин формулу:
. Несколько ранее (1881 г.) Чамичан (Ciamician) и Деннштедт дали Пиридин формулу:  . Бамбергер и Пехманн в 1891 г. предложили центрическую формулу. Все эти формулы имеют многое за себя, но в то же время ни одна из них не удовлетворяет вполне всем существующим фактам. Интересно то обстоятельство, что Томсен, основываясь на молекулярной теплоте сгорания Пиридин, приходит к заключению, что в Пиридин не имеется двойных связей и, таким образом, как бы подтверждаются формулы Бамбергера и Пехманна и Чамичана и Деннштедта [Относительно приложения физических и физико-химических методов к исследованию вопросов химического строения — см. Строение химическое.]. Несмотря на неполную выясненность строения Пиридин и его производных, химия этих соединений весьма успешно разрабатывается в других отношениях. К этому привлекает главным образом то обстоятельство, что многие алкалоиды принадлежат к дериватам рассматриваемого класса соединений. В 1879 г. А. Вышнеградский высказал мнение, что, может быть, все растительные основания суть производные Пиридин или хинолина (см.), а в 1880 г. Кенигс предлагал даже именем алкалоидов называть только те растительные основания, которые могут быть рассматриваемы, как дериваты Пиридин Из строения Пиридин кольца очевидно, что теоретически возможны 3 изомерных однозамещенных Пиридин, 10 двузамещенных и т. д. Углеродные атомы кольца обозначаются греческими буквами или цифрами по приведенной схеме, которые и ставятся перед названием соответствующей боковой цепи. Самое кольцо часто в эмпирических формулах обозначается слогом Ру
. Бамбергер и Пехманн в 1891 г. предложили центрическую формулу. Все эти формулы имеют многое за себя, но в то же время ни одна из них не удовлетворяет вполне всем существующим фактам. Интересно то обстоятельство, что Томсен, основываясь на молекулярной теплоте сгорания Пиридин, приходит к заключению, что в Пиридин не имеется двойных связей и, таким образом, как бы подтверждаются формулы Бамбергера и Пехманна и Чамичана и Деннштедта [Относительно приложения физических и физико-химических методов к исследованию вопросов химического строения — см. Строение химическое.]. Несмотря на неполную выясненность строения Пиридин и его производных, химия этих соединений весьма успешно разрабатывается в других отношениях. К этому привлекает главным образом то обстоятельство, что многие алкалоиды принадлежат к дериватам рассматриваемого класса соединений. В 1879 г. А. Вышнеградский высказал мнение, что, может быть, все растительные основания суть производные Пиридин или хинолина (см.), а в 1880 г. Кенигс предлагал даже именем алкалоидов называть только те растительные основания, которые могут быть рассматриваемы, как дериваты Пиридин Из строения Пиридин кольца очевидно, что теоретически возможны 3 изомерных однозамещенных Пиридин, 10 двузамещенных и т. д. Углеродные атомы кольца обозначаются греческими буквами или цифрами по приведенной схеме, которые и ставятся перед названием соответствующей боковой цепи. Самое кольцо часто в эмпирических формулах обозначается слогом Ру  . Что касается вопросов об определении взаимного положения боковых цепей производных Пиридин, то очевидно, что вопросы эти могли быть разрешаемы только тогда, когда были точно установлены положения боковых цепей таких однозамещенных Пиридин, к которым в большинстве случаев легко было бы перейти от изучаемого производного. Такими однозамещенными производными являются пиридинкарбоновые кислоты. Благодаря трудам Скраупа и Ладенбурга было вполне установлено, что пиколиновая кислота есть α-пиридинкарбоновая, никотиновая — β-Пиридин-карбоновая, а изоникотиновая — γ-Пиридин-карбоновая кислота.
. Что касается вопросов об определении взаимного положения боковых цепей производных Пиридин, то очевидно, что вопросы эти могли быть разрешаемы только тогда, когда были точно установлены положения боковых цепей таких однозамещенных Пиридин, к которым в большинстве случаев легко было бы перейти от изучаемого производного. Такими однозамещенными производными являются пиридинкарбоновые кислоты. Благодаря трудам Скраупа и Ладенбурга было вполне установлено, что пиколиновая кислота есть α-пиридинкарбоновая, никотиновая — β-Пиридин-карбоновая, а изоникотиновая — γ-Пиридин-карбоновая кислота.
Пиридин C5H5 N, первый член гомологического ряда пиридиновых оснований, находится в костяном масле, каменноугольной смоле, аммиачной воде газового производства, в продуктах разложения нек. алкалоидов и в продажном амиловом спирте. Синтетического метода получения Пиридин, дающего сколько-нибудь хорошие выходы его, до сих пор не найдено; в теоретическом же отношении интересны синтезы Чамичана и Деннштедта:
I. C4H4 HK (пиррол-калий) + СНСl 3 = C5H4 ClN (хлорпиридин) + KCl + НСl
II. С 4 Н 4 NК + CH 2J2 = C5H5N + KJ + JH.
Вполне чистый Пиридин легче всего получается из сырого продажного продукта, подвергая его действию окислителей; тогда гомологи Пиридин дают карбоновые кислоты, Пиридин же остается неизмененным и может быть отделен от кислот промыванием едкими щелочами; продажный продукт можно также перевести в хлористоводородную соль и осаждать Пиридин из слабых солянокислых растворов в виде двойной соли с HgCl 2 или с ферроциановокалиевой солью. Очищенный тем или другим способом Пиридин представляет бесцветное, не изменяющееся на воздухе масло с неприятным запахом, кипящее при 116—117°, уд. веса 1,0033, растворимое в воде, спирте, эфире и т. д. Пиридин обладает ясно выраженным характером одноатомного основания; соли его легко растворимы в воде и образуют многочисленные двойные соли. С водой Пиридин образует кипящий при 92—93° гидрат C 5H5N·3H2 O. Чистый Пиридин не реагирует с самыми сильными окислителями: в этом отношении его кольцо прочнее бензольного; наоборот, от действия HJ при 300° Пиридин разрушается, давая аммиак и пентан. По аналогии с бензолом нужно было бы ждать образования пентаметилена или пентаметиленамина. При восстановлении натрием в спиртовом растворе Пиридин переходит в гексагидро-Пиридин, или пиперидин (см. ниже); при действии же натрия на сухое основание получается 4-дипиридил C 5H4N—C5H4 N, дипириден C 10H10 N и изоникотин C 10H14N2; первый продукт образуется также при пропускании паров Пиридин через раскаленные трубки. Пиридин, имея в своем ядре третичный азот, способен соединяться с йодюрами спиртов, давая аммониевого типа пиридиниевые соединения, напр., Пиридин с йодистым метилом дает C 5H5N—CH3 —J; эти соединения тверды, часто аморфны, с влажной окисью серебра дают малопрочные, сильно щелочные основания, которые при перегонке с едким кали переходят в замещенные дигидро-Пиридин, а при нагревании до 290—300° в замещенные Пиридин При нагревании с монохлороуксусной кислотой Пиридин дает хлористоводородный Пиридин-бетаин:
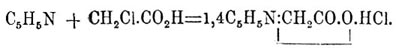
Гомологи Пиридин получаются: 1) при нагревании альдегидаммиаков с альдегидами или кетонами:
CH3—CHO—NH3 + 3CH3CHO = C5H3N(CH3)(I)—(C2H5)(VI) + 4H2O;
3CH3—CHO—NH3 + 6(CH3)2CO = 2C5H2N(CH3)3(II, IV, VI) + C5H3N(CH3)3(II, IV, VI) + 9H2O;
2) нагреванием йодалкильных производных Пиридин до 290—300°; по этой реакции получаются главным образом α- и γ-гомологи:
C5H5N—JCH3 = С 5 Н 4 (СН 3)N—HJ;
3) нагреванием α-пиридиновых оснований с альдегидами или кетонами до 250—260°:
С 5 Н 4 N—СН 3 + СН 3 СНО = С 5 Н 4 N—СН=СН—СН 3 + Н 2 О;
С 5 Н 4 N—СН 3 + CO(CН 3)2 = С 5 Н 4 N—СН=C(СН 3)2 + Н 2 О;
4) разложением пиридинкарбоновых кислот при нагревании или перегонке с известью:
С 5 Н 4N—CO2 H = С 5 Н 5 N + СО 2;
5) из костяного масла (ol. animal. Dippeli) дробной перегонкой (Андерсон, Вейдель, Ладенбург и Рот, Чамичан) [Чамичан совместно с Вейделем показал, что присутствие пиридиновых оснований в костяном масле нужно приписать взаимодействию, совершающемуся при высокой температуре сухой перегонки костей между высшими жирными альдегидами и аммиаком; эти же продукты образуются, с одной стороны, разложением кислот жира, а с другой, при сухой перегонке клея, так как чистый клей (желатин) при этих условиях дает аммиак, жирные амины и пиррольные производные. На основании этого в костяном масле, приготовленном из обезжиренных костей, не может находиться Пиридин]. Теоретически их можно образовать, замещая последовательно водороды пиридинового ядра углеводородными радикалами. Благодаря гетерогенности ядра число изомеров пиридиновых оснований весьма велико, и далеко не все из них получены. Теория предсказывает 3 метилпиридина C 5H4N—CH3; все они получены и известны под названием пиколинов. В 1846 г. Андерсон нашел в каменноугольной смоле основание C 6H7 N, которое и назвал пиколином от слов pix — смола и oleum — масло. Позднее то же основание было выделено из продуктов сухой перегонки битуминозных сланцев и некоторых сортов торфа, из продуктов разложения цинхонина (при перегонке его с едким кали) и паров никотина (при прохождении их через раскаленные трубки). Исследование этого пиколина показало, что он состоит главным образом из 2 изомеров: α-метил-Пиридин, или α-пиколина, и β-метил-Пиридин, или β-пиколина. α-Пиколин впервые был выделен Вейделем в 1879 г. из сырого пиколина при помощи двойной платиновой соли. α-Пиридин представляет бесцветное, легко растворимое в воде масло, кипящее при 129—130°, уд. в. 0,965. β - Пиколин открыт Байером в 1870 г. при сухой перегонке акролеин-аммиака. Получается он обыкновенно при нагревании глицерина с фосфорным ангидридом и фосфорноаммиачной солью. β-Пиколин по своим свойствам весьма похож на α-пиколин, кипит при 142—143°, уд. вес D°о = 0,977. γ - Пиколин открыт Вейделем в сыром пиколине и получается при перегонке изоникотиновой кислоты и спартеина. Точка кипения его 144—145°, уд. вес D = 0,971—0,974. Как видно из сопоставления вышеприведенных данных, все три изомерных метил-Пиридин весьма близки между собой по своим физическим свойствам; та же близость наблюдается и в их химическом характере; кроме свойств, присущих Пиридин, пиколины обладают следующими чертами, специфическими для гомологов Пиридин: 1) действием окислителей они легко переходят в пиридинкарбоновые кислоты C 5H4N—CH3 + 3О = C 5H4 N—CО 2 —Н + Н 2 О; 2) при нагревании с альдегидами или кетонами они образуют или гомологи Пиридин с непредельной боковой цепью, или же оксипиридиновые основания. Так, при нагревании с бензойным альдегидом *-пиколин дает *-стильбазол C5H4N.CH:CH.C6H5, кристаллическое вещество, плавящееся при 90, 5° — 91° и кипящее при 324 — 325°. Следующие за пиколинами — гомологи Пиридин известны под названием лутидинов. Название это дано было в 1851 г. Андерсоном основанию, выделенному им из костяного масла и имеющему формулу С 7 Н 9 N, тождественную с формулой толуидина. Для указания на это тождество Андерсон и предложил название "лутидин", полученное перестановкой букв в слове "толуидин". Впоследствии оказалось, что лутидин Андерсона есть смесь различных оснований. Теория предсказывает 9 возможных изомерных лутидинов, из которых получены только 8, именно: 1) α -, β - и γ -этил-Пиридин получены синтетически и, кроме того, β - и γ - этил.-Пиридин находятся в продуктах разложения цинхонина, стрихнина и бруцина; 2) пять диметилпиридинов: αα ' лутидин, или αα'-диметилпиридин, выделен в чистом состоянии в 1885 г. Ладенбургом из каменноугольной смолы, представляет жидкость с мятным запахом, более растворимую в холодной воде, чем в горячей (черта, свойственная всем дальнейшим гомологам), кипящую при 142°, уд. в. D° 0 = 0,9424. αγ -Лутидин, полученный Ладенбургом одновременно с αα'-лутидином, — жидкость с запахом свежих огурцов, кипит при 156—157°, уд. в. D° 0 = 0,9503. ββ ' -Лутидин получен в 1890 г. Дюркопфом и Геттшем перегонкой ββ'-метилпиколиновой кислоты с известью, представляет сильно преломляющую свет жидкость с приятным запахом, кипящую при 169—170°, уд. в. D°0 =0,9614. Относительно четвертого изомера αβ'-лутидина известно только, что он кипит при 162—166° и при окислении дает изоцинхомероновую кислоту. βγ-Лутидин исследован в 1896 г. Аренсом, получается из каменноугольной смолы, кипит при 163,5°—164,5°. Коллидины C8H11 N. Название дано Андерсоном, как основанию, изомерному ксилидину. Теоретически возможны 22 изомера: 3 пропил-Пиридин, 3 изопропил-Пиридин, 6 триметил-Пиридин и 10 метилэтил-Пиридин Получено только 11 с известной группировкой боковых цепей и 4 с неизвестной. Из них наиболее интересны: α-пропил-Пиридин, или конирин, получен впервые Гофманном в 1881 г. при перегонке с цинковой пылью хлористоводородного кониина, жидок, кипит при 165—168°, легче воды, обладает неприятным, напоминающим Пиридин запахом; α-метил-β'-этил-Пиридин, альдегидколлиоин, альдегидин, находится в сивушном масле; ароматическое масло, кипит при 173—174°, уд. в. D° 0 = 0, 9395; αα ' γ -триметил-Пиридин, симметрический коллидин, жидок, кип. при 171—172°, уд. в. D° 0 = 0, 9312. Парволин C9H13 N (название дано Вильямсом основанию по причине его малой летучести) выделен из продуктов сухой перегонки битуминозных сланцев Дорзетшайра; из 54 возможных изомерных парволинов известны только 11, из которых 6 с неизвестным строением: αβγβ ' -тетраметил-Пиридин выделен Аренсом из каменноугольной смолы, жидок, кипит при 233—234°. Фенилированные Пиридин имеют только исторический интерес, так как α- и β -фенил-Пиридин C5H4N—C6H5 послужили Скраупу и Кобенцелю для установки формулы строения пиколиновой и никотиновой кислот (см. ниже).
Гидропиридины — так называются продукты гидрогенизации Пиридин и его гомологов; теоретически возможны ди-, тетра- и гексагидро-Пиридин, из них только последние более или менее полно изучены, первые же два типа почти неизвестны. Гексагидро-Пиридин, или пиперидины, — весьма важные соединения в химии алкалоидов. Наилучшим способом их получения нужно считать восстановление металлическим натрием в спиртовом растворе соответствующих Пиридин: С 5 Н 5 N + 3Н 2 = С 5 Н 10 NН; однако они получаются и конденсацией хлористоводородных солей жирных диаминов (см. Имины). Как Пиридин по прочности своего кольца приближается к бензолу и, следовательно, вообще к ароматическим соединениям; так, пиперидин и его гомологи, будучи вполне насыщенными соединениями, имеют характер жирных соединений. Кольцо пиперидинов легко разрывается при действии окислителей, в большинстве случаев с выделением аммиака и образованием жирных кислот; иногда же при простой перегонке Пиридин соединения выделяется вода и жирный амин. Подобно всем иминам, пиперидины дают нитрозосоединения и присоединяют йодистые спиртовые радикалы. В этом случае особенно поразительна способность пиперидина присоединять бром- и йод-бензолы, давая прочные п-фенилпиперидины. Эта способность в гомологах пиперидина значительно ослаблена. Пиперидин С 5 Н 10 NН, простейший член этого ряда соединений, представляет жидкость с перечным запахом, кипящую при 106°. Открыт Кагуром в продуктах перегонки пиперина (см.) с натристой известью. Синтетически может быть получен, кроме указанных выше общих реакций, нагреванием хлор- или бромалкиламина с раствором едкого кали:
NН 2 (СН 2)5 —Вr + КНО = C 5H10 —NH + KBr + Н 2O.
При действии HJ при высокой температуре пиперидин распадается на аммиак и пентан. Перекись водорода весьма характерно реагирует с пиперидином; при этом наблюдается размыкание кольца с образованием альдегида амидовалериановой кислоты:
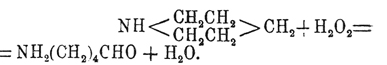
Рядом с этим происходит простое окисление двух групп СН 2 в СО, результатом которого получается имид (см.) глутаровой кислоты. Как всякий вторичный амин, пиперидин реагирует с ангидридами или хлорангидридами кислот, и получаемые замещенные пиперидины весьма легко окисляются, образуя производные амидомасляной или амидовалериановой кислоты. При действии 2 мол. йодистого метила пиперидин дает йодистый диметилпиперидиний; эта соль с влажной окисью серебра переходит в свободное основание С 5 Н 10 N(ОH)(СН 3)2, которое настолько непрочно, что при простой перегонке его выделяется вода и разрывается кольцо с образованием пентенилдиметиламина (СН 3)2 N(СН 2)3 —СН=СН 2, который раньше неправильно назывался диметилпиперидином. Этот последний продукт также способен реагировать с CH 3 J и Ag 2 O, давая гидрат окиси пентенилтриметиламмония (OH)(CH3)3N(CH2)3—CH=CH2, который, в свою очередь, при перегонке разлагается на воду, триметиламин и пиперилен СН 2 =СН—СН 2 —СН=СН 2. Гомологи пиперидина повторяют свойства самого пиперидина и до сих пор не представляют большого научного интереса. Названия их производятся вставкой слога "пе" после первого слога названия соответствующего пиридинового основания, след., рядовые их названия будут: пипеколины, лупетидины, копеллидины и т. д. Из этих соединений имеет интерес только α -пропилпиперидин, как тождественный с алкалоидом кониином (см.).
Непосредственное замещение водородов ядра отрицательными радикалами в Пиридин идет вообще крайне трудно; галоидозамещенные Пиридин получаются только при действии галоидных соединений фосфора или сурьмы на окси-Пиридин, при непосредственном же хлорировании выходы ничтожны, а в случае гомологов Пиридин галоид идет в боковые цепи. При действии PCl 5 на Пиридин получается тетрахлор- и пентахлор-Пиридин Сульфация идет несколько легче, и при весьма продолжительном кипячении Пиридин с крепкой серной кисл. получается β-сульфопиридиновая кислота C5H4NSO3 H, которая довольно легко обменивает свою сульфогруппу на другие отрицательные радикалы и, напр., с цианистым калием дает β-циан-Пиридин, или нитрил никотиновой кислоты C 5H4 N—CN, плавящийся при 49°. Нитрация удается только тогда, когда пиридиновое ядро имеет группы NH 2, или ОН, или др., облегчающие нитрование и бензольного ядра; но и в этих случаях приходится прибегать к продолжительному нагреванию или дымящейся азотной кислоте. Ввиду только что указанных обстоятельств нитро-Пиридин неизвестны, получены же только оксинитро- и амидонитропроизводные. Амидопиридины получаются исключительно по реакции Гофманна, обработкой амидов Пиридин-карбоновых кислот бромом в щелочном растворе. Амидо-Пиридин являются высококипящими твердыми телами, мало изменяющимися на воздухе.
Оксипиридины по своему химическому характеру отвечают амидофенолам; они соединяются, давая соли, как со щелочами, так и с кислотами, причем кислотность их возрастает с возрастанием числа гидроксильных групп, а основные свойства падают, и уже триокси-Пиридин с кислотами образуют соли, легко разлагающиеся водой. Получаются окси-Пиридин из соответствующих окси-Пиридин-карбоновых кислот, которые с необыкновенной легкостью и количественно выделяют СО 2 из своего карбоксила. Большая часть окси-Пиридин хлорным железом окрашивается в красный цвет. Кроме вышеуказанного двойственного характера, α- и γ-окси-Пиридин обладают еще функцией кетонов, и поэтому их можно рассматривать как кето- или оксопроизводные гидропиридинов, почему их и называют иногда пиридонами и, следов., α- и γ-окси-Пиридин могут изображаться двумя рациональными формулами:
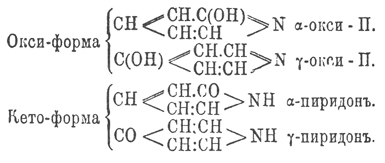
Здесь приходится столкнуться с очень интересным случаем таутомерии: по-видимому, группировки — CO—NH— и —C(OH)=N — вообще очень непостоянны и легко переходят одна в другую под влиянием различных реагентов; даже в таких соединениях, как кислотные амиды R—CONH 2, замечается тенденция образовать производные имида R—C(OH)=NH, в циклических же соединениях, подобных α- или γ-окси-Пиридин, эта способность всегда весьма резко выражается в производных, тогда как начальное вещество, по-видимому, не имеет двух форм и потому неизвестно, какую из них нужно придавать ему. Разбираемый здесь случай таутомерии (см.) еще интересен и тем, что с ясностью указывает на таутомерию и самого пиридинового кольца в α- и γ-пиридонах: из них первый отвчает формуле Кернера, а второй — формуле Риделя. Вообще, причина появления таутомерных форм не выяснена еще в достаточной степени; однако с некоторым вероятием здесь можно предполагать влияние температуры, с повышением которой ослабляется постоянство связей и, след., причина таутомерии; по этому воззрению, высказанному в последнее время Кнорром, есть как бы начало, или первая степень, диссоциации. Придерживаясь этого взгляда, можно думать, что, выделяя, напр., α-окси-Пиридин при весьма низкой температуре один раз из α-этоксипиридина С 5 Н 4 (ОС 2 Н 5)N, а другой раз из п - этил-α-пиридона C5H4ON—C2H5 мы получим два различных вещества. α-Окси-пиридин C5H5 ON, твердое тело с точкой плавления 106°, получается из оксиникотиновой и оксихинолиновой кислот; с бромной водой дает дибромокси-Пиридин С 5 Н 3 Вr 2 ОN. При обработке его йодистым этилом получается п-этил - α-пиридон C5H4ON—C2H5, кипящий при 247°, при действии же иодистого этила на серебряную соль α-окси-Пиридин получается этильный эфир α-окси-Пиридин, кипящий при 156°. β-Окси-Пиридин получается при сплавлении β-сульфопиридиновой кислоты с едким кали, плавится при 154° и кипит без разложения. γ-Окси-Пиридин получается из оксипиколиновой кислоты, плавится при 146°; п-метил- γ-пиридон плавится при 89°, а γ-метокси-Пиридин жидок и кипит при 190°. Диоксипиридины C5H3(OH)2 N получаются из диокси-Пиридин-карбоновых кислот и из них наиболее интересны отвечающие формуле R (β)—C5H2(OH)(α, α ') —N, так как по расположению гидроксилов они напоминают резорцин и с фталевым ангидридом (см.) дают краски.
Пиридилалкины теоретически производятся из гомологов Пиридин замещением атомов водорода боковых групп оксигруппой, след., рядом с чертами, свойственными Пиридин, они обладают еще и характером спиртов. Получаются они: 1)конденсацией α-метилированных Пиридин с альдегидами, при нагревании с водой: Pу—CН 3 + CH2 =O = Pу—CH 2CH2OH; Py—CH3 + О=СН—ОН 3 = РуСН 2 СН(ОН)—СН 3; 2) из гомологов Пиридин, бромированных в боковой цепи: Ру—СН 2 Вr + КНО = РуСН 2 (ОН) + КВr; 3) воcстановлением пиридилкетонов: Ру—СОС 2 Н 5 + Н 2 = Ру—СН(ОН)—С 2 Н 2. Пиридилалкины суть жидкости, кипящие выше 200°. Представителем может служить α - пиколилалкин РуСН 2 СН 2 ОН, кипящий при 179° под давлением в 22 мм.
Пиридилкетоны. Этим именем в отличие от пиридонов называются такие производные, которые имеют группу СО не в ядре, а в боковой цепи, причем до сих пор известны только пиридилкетоны типа Py—CO—R. Получаются они перегонкой кальциевых солей Пиридин-карбоновых и соответствующих жирных кислот (Ру—СО 2)2 Са + (СН 3 СО 2)2 Са = 2Py—COCH 3 + 2CaCO3 (ср. Кетоны). При восстановлении, подобно жирным кетонам, они дают вторичные спирты (см. выше). Все они жидки, кипят выше 200° без разложения. α-Пиридилэтилкетон α -PyCOC2H5, получающийся перегонкой кальцевых солей пиколиновой и пропионовой кислот, кипит при 205°, натрием в спиртовом растворе восстановляется в α - этилпиперилалкин C2H5CH(OH)—C5H9 —NH, тождественный с алкалоидом псевдоконгидрином.
Пиридинкарбоновые кислоты получаются окислением гомологов Пиридин раствором марганцево-калиевой соли, причем все боковые группы, как жирные, так и ароматические, сгорают до карбоксилов. По этой причине большая часть алкалоидов, как производные Пиридин, при энергичном окислении в виде конечных продуктов дают Пиридин-карбоновые кислоты. Синтетический метод получения Пиридин-карбоновых кислот, выработанный Гантчем, основан на конденсации ацетоуксусного эфира с альдегидоаммиаками и разбивается на две части: на получение дигидро-Пиридин-дикарбонового эфира и окисление этого последнего: 2CH 3 —CO—СН 2—CO2C2H5 + CH3—CH(OH)NH2 = С 5 Н 2 (СН 3)3(CO2C2H5)2 N + 3Н 2 О; С 5 Н 2 (СН 3)3(CO2—C2H5)2 N + О = С 5 (СН 3)3(CO2—C2H5)2 N + Н 2 О. При этом Гантч и Эпштейн показали, что всегда в этом случае метильныt группы ацетоуксусного эaира становятся в α -, α'- положения, а радикал альдегида в γ-положение. Кроме указанных способов, монокарбоновые кислоты могут быть получены из поликарбоновых, которые при нагревании легко выделяют СО 2 из своих карбоксилов, причем обыкновенно наблюдается первоначально отщепление CO 2 из α-положения. Пиридин-карбоновые кислоты по своему химическому характеру напоминают амидокислоты, причем основные свойства этих последних в них постепенно исчезают с накоплением карбоксильных групп. Натрием в спиртовом растворе они, подобно всем пиридиновым соединениям, восстановляются с присоединением атомов водорода, переходя в соответствующие пиперидинкарбоновые кислоты. В Пиридин-карбоновых кислотах, вообще говоря, Пиридин ядро сильно ослаблено, так как весьма многие Пиридин-карбоновые кислоты при действии амальгамы натрия в щелочном спиртово-водном растворе восстановляются с выделением NH 3 в жирные окси- или лактонокислоты; при этом группировка —CH=N—CH= переходит в CO 2H(NH2)CH(OH)— или в —СО—О—СН 2 и (NН 3), например:
α C5H4N—CO2H + H2 + 3Н 2 О = СО 2 Н—СН 2 —СН 2 —СН 2 —СH(ОН)—СО 5 Н + NH 3 и
СО 2 Н—СН 2 —СН 2 —СН 2 —СH(ОН)—CO 2 H — Н 2 О = 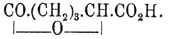
В теоретическом отношении Пиридин-карбоновые кислоты важны потому, что они служат базами для суждения о расположении боковых групп различных Пиридин соединений, которые, как видно из вышесказанного, окисляясь, легко в них переходят. По этой причине строение их весьма тщательно разрабатывалось Скраупом, Ладенбургом и др., которые и установили вполне точно положение карбоксильных групп относительно атома азота. Пиридинмонокарбоновые кислоты: Пиколиновая кислота α-С 5 Н 4 N—СО 2 Н плавится при 135—136° и возгоняется. Получается окислением α-пиколина (Вейдель) и, как все α-Пиридин-карбоновые кислоты, раствором железного купороса окрашивается в желтый цвет. Никотиновая кислота β -C5H4N—CO2 H, темп. плав. 209°, получается окислением β-пиколина, открыта Губером при окислении никотина (см.) хромовой смесью. Изоникотиновая кислота γ -C5H4N.CO2 H плавится при 304°, получается окислением γ-пиколина и отщеплением углекислоты от цинхомероновой кислоты (Скрауп и Гугеверф, ван-Дорп). Пиридиндикарбоновые кислоты. Хинолиновая кислота αβ-С 5 Н 3 N(СО 2 Н) 2, темп. пл. 190°, получается окислением хинолина (см.) и его замещенных (в бензольном ядре) производных. Цинхомероновая кислота β, γ-С 5 Н 3 N(СО 2 Н) 2, темп. пл. 266°, получается окислением цинхонина, цинхонидина или изохинолина, при восстановлении амальгамой натрия легко переходит в цинхоновую кислоту С 7 Н 6 О 5, а эта последняя при нагревании распадается на СО 2 и пироцинхоновую кислоту или ангидрид диметилмалеиновой кислоты. Лутидиновая кислота * α , γ -C5H3N(CO2H)2 + 2H2 O, темп. пл. 235°. Изоцинхомероновая кислота αβ '-C5H3N(CO2H)2, темп. пл. 236°. Дальнейшие Пиридин-поликарбоновые кислоты мало изучены; некоторый интерес из них представляют бербероновая кислота α ' βγ -C5H2N(CO2H)3, получающаяся окислением алкалоида берберина, и Пиридин-пентакарбоновая кисл. С 5 N(СО 2 Н) 5 + 2Н 2 О, получающаяся окислением коллидиндикарбоновой кислоты. Оксипиридинкарбоновые кислоты важны как исходные продукты для получения оксипиридинов (см. выше). В своих производных они, как и окси-Пиридин, являются в 2 таутомерных формах — или как окси-, или же как кетосоединения. По своему характеру эти кислоты напоминают амидоокси- или амидокетокислоты и получаются особенно легко при действии аммиака на соответствующие пиронкарбоновые кислоты (см. Пирон): С 5 Н 3 О 2—CO2H + NH3 = C5H4(OH)N—CO2H + H2O.
Д. А. Хардин
|
Смотрии так же... |
|
|